
Site Menu

To top
This help page provides detailed descriptions of the different steps in the ConTra analysis process
| Click on a topic below for more information: |
| Step 1 - Step 2 - Step 3 - Step 4 - Results - Exploration |
| Examples - Demos - FAQs |
| Step 1: type of analysis and your input data | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
ConTra can perform two types of analyses depending on the question of the researcher (1).
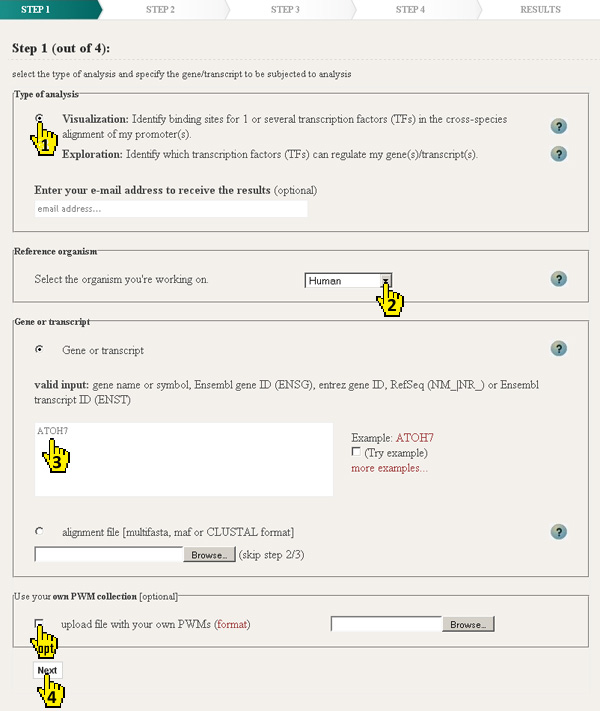
Since version 2 of ConTra (ConTrav2) it is possible to choose your reference organism (2)
To specify your gene of interest (3) ConTra accepts the official name or symbol (HGNC,
Entrez Gene),
aliases or gene identifiers (Entrez Gene),
RefSeq accession numbers (NM_,NR_) or
Ensembl accession numbers (ENSG, ENST).
If the gene or transcript is relatively new and/or no multiz alignment is availble, it is possible to upload your own multiple alignment file (MAF). ConTra accepts MAFs in the UCSC maf format (example.maf), clustal format (example2.aln) or fasta format (example3.fasta). When a maf file is uploaded, step 2 and 3 will be skipped. It is also possible to upload your own collection of positional weight matrices (PWMs) (optional). This collection will be used next to the built-in PWM libraries from JASPAR, TRANSFAC, phyloFACTS and PBM homeodomains. |
| Step 2: a transcript of your gene of interest |
|
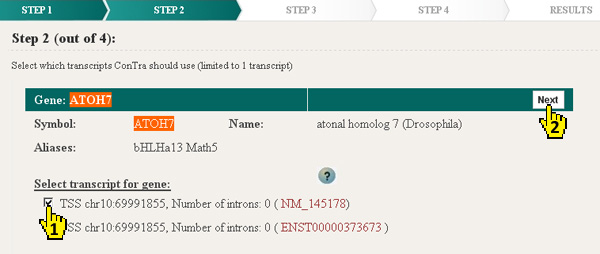
ConTra shows a list of genes that contain the name, symbol or identifier provided in the first step. The matching keyword is highlighted. A gene can encode several transcipts or isoforms. These can be regulated differently by the presence or absence of specific transcription factor binding sites in the transcript specific promoter, UTR or intron region. For every transcript that can be analyzed in ConTra the position of the transcription start site (TSS) is shown together with the number of introns and the identifier (RefSeq of Ensembl). The identifier is linked to the genomic view of this transcript in the UCSC or Ensembl genome browser.
|
| Step 3: promoter, 5'UTR, 3'UTR or any intron |
|
In the first version of ConTra it was only possible to analyze the promoter upstream of the TSS. In the new version ConTra v2 next to the promoter region also 5' UTR, 3'UTR or any intron can be analyzed (1). The promoter size upstream of the TSS can be specified. For UTRs and introns the entire region is being used in the analysis. After selecting the sequence parts clicking Next will proceed to the final step 4 (2). 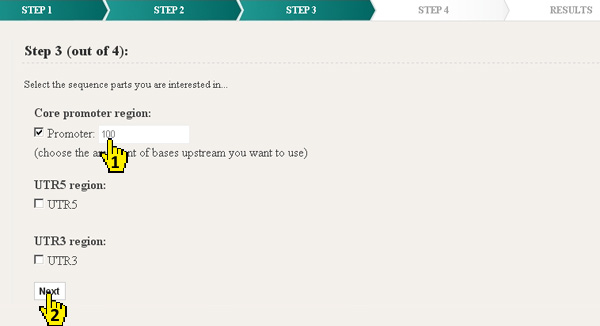
|
| Step 4: transcription factor bindings sites (TFBS) and stringency |
|
One of four stringencies can be selected next to the "minimize false positives" option (1). This last option only works for PWMs selected in the TRANSFAC list. The different stringencies balance sensitivity and accuracy. With a core match = 0.85 and a matrix match = 0.70 the detection will be highly sensitive but less accurate and some false positives may be included. Consequently a core match = 1.00 and matrix match = 0.95 will be very accurate but less sensitive and may not show some true binding sites.
Position weight matrices or PWMs represent sequence motifs for transcription factor binding sites (TFBS). 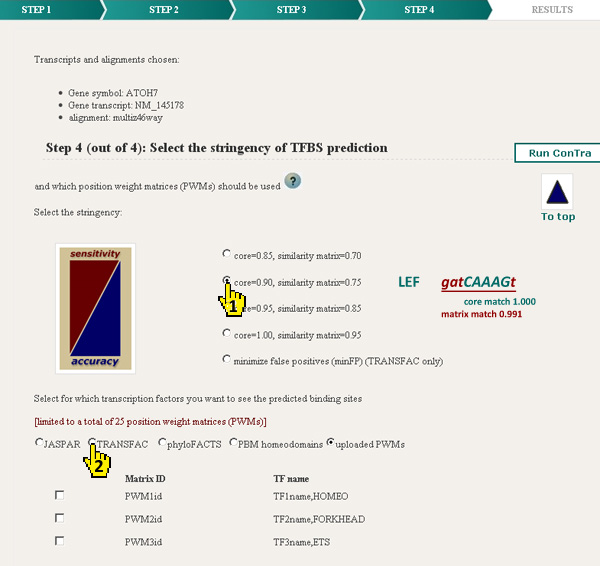
Up to 25 PWMs can be selected from the different databases (3). The lists of available PWMs are sorted alphabetically. 
|
| Results |
|
Depending on the size of the sequence parts (length of the sequences to scan), 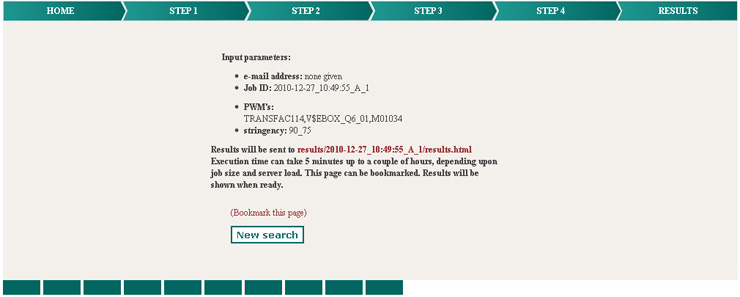
The result page shows results for every sequence part (promoter, UTRs, introns or own uploaded maf). The multiz alignments are divided in alignment blocks. For every block a jalview preview figure is shown and if TFBS were detected these are highlighted (1). 
On the html page of a specific block the different PWMs can be visualized or hidden by clicking the checkboxes. Some binding sites might overlap and can only be partially visible. The binding sites corresponding with the first PWM in the list will be the most in the back (bottom layer). The last one in the list will be on top (front layer). Deselecting the checkboxes of the sites shown in the front will reveal the underlaying sites as illustrated in the example below. 
|
| Exploration |
|
The exploration option helps the scientist who has no idea which transcription factors (TFs) may regulate his or her gene of interest. This analysis consists of the same 4 steps: selecting the gene of interest, selecting the transcript, selecting the sequence parts (promoter region, UTRs, introns) and selecting the PWMs. Step 4 is somewhat different than in the visualization process. Instead of specifying some PWMs the user has to select the databases to be used. The alignment will be analyzed by all PWMs from the selected databases and will therefore need a much longer execution time. For running one set of PWMs on the human reference multiz46way alignment the server will typically need about half an hour. 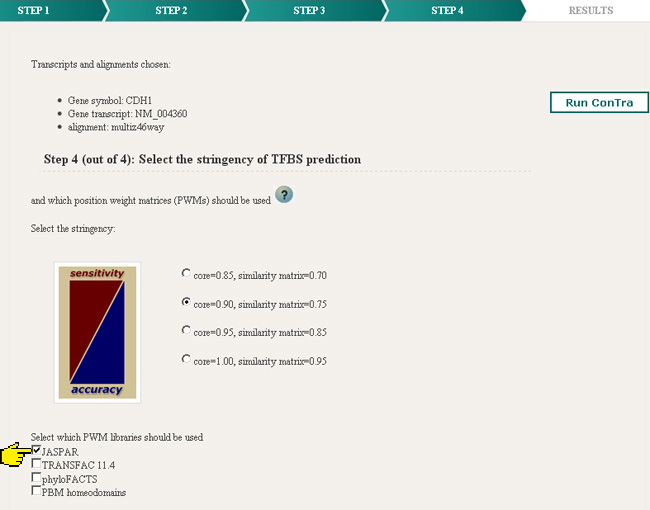
After the initial analysis ConTra will show a list of the putative TFBS ranked by their binding probability to the regio of interest. By default the top 20 is listed. Click expand to show the top 100. At the bottom of the first result page there is also a link to the rankedlist file which contains the results of all PWMs from the selected databases. 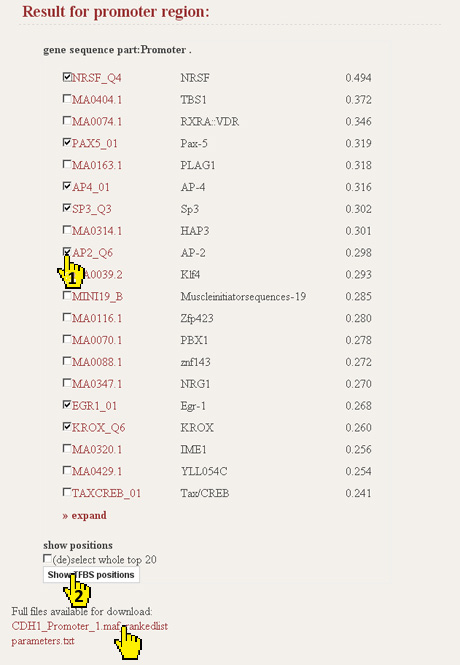
|
| Examples | ||||||||
|

| FAQs |
|
Q: No transcripts were found by your query! ConTra cannot find your gene of interest.
Q: We are sorry but UCSC genome has no correct maf available for the selected position.
Q: The uploaded file is not a text file! For other questions or help contact us by sending an email to ConTra@irc.UGent.be |
| New search | Help | Contact |